Voltage- and time-dependent valence state transition in cobalt oxide catalysts during the oxygen evolution reaction
The oxygen evolution reaction (OER) is the rate-limiting step for renewable-energy technologies, such as direct solar, water splitting, rechargeable metal-air batteries and regenerative fuel cells to meet the requirement of rapidly increasing global energy demand. To make improvements, the complex reaction processes and the catalytic active sites have to be determined. There is especially a high need to do this determination during the electrochemical reaction itself, i.e. in-operando.
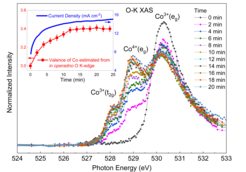
The team around Zhiwei Hu from the MPI CPfS, together with colleagues from the Shanghai Institute of Applied Physics of the Chinese Academy of Sciences, the Beijing University of Chinese Academy of Sciences, the Tamkang University and the National Synchrotron Radiation Research Center in Taiwan, have successfully applied soft X-ray absorption spectroscopy (XAS) to follow in operando the valence and spin state of the Co ions in Li2Co2O4 under oxygen evolution reaction (OER) conditions. They have observed that a substantial fraction of the Co ions undergo a voltage- and time-dependent valence state transition from Co3+ to Co4+ together with a spontaneous delithiation. Fig. 1 shows that the Co4+ related spectral weight and the current density j (inset) increase with the time under applied voltages of 1.6 V. It is also remarkable that the edge-shared Co–O network and spin state of the Co ions remain unchanged. Density functional theory calculations confirm that the highly oxidized Co4+site is mainly responsible for the high OER activity, rather than the intermediate spin state Co3+ or the oxygen vacancy sites as often thought. The complex many-body electronic structure of the system is displayed by the cartoon in Fig.2, highlighting the importance of the oxygen ligand hole states around the Co4+ ions.
This work has been featured in a Nature Communications Editors’ Highlights webpage of recent research on Energy Materials.

 is marked by a dashed vertical line. Considering that the position of the Ef is arbitrary in the band gap, we use the broad O 2p band as energy reference. The Ef then shifts towards to the center O 2p band with increasing Co valence.")
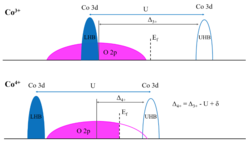
Fig. 2 The schematic electron removal and addition diagram of Co3+ and Co4+-based oxides. Broad O 2p bands are shown in magenta and narrow Co 3d bands in blue. The Fermi level (Ef) is marked by a dashed vertical line. Considering that the position of the Ef is arbitrary in the band gap, we use the broad O 2p band as energy reference. The Ef then shifts towards to the center O 2p band with increasing Co valence.
ZH/CPfS