Composition of clathrates: Computational structure analysis
The prediction of crystal structures and the optimisation of their atomic positions has become an important tool in material science. However, standard calculations have important limitations. For example, the atomic arrangement is usually calculated using a 0 K approach without taking into account entropy. Moreover, solutions are often restricted to a given symmetry while atomic defects are neglected. In the case of semiconductors used e.g. in the thermoelectric energy conversion, understanding of structural details is highly important. In a collaboration between the Fritz Haber Institute in Berlin and the department Chemical Metals Science at MPI CPfS, complex structural features of clathrate phases have been verified theoretically. By using the FHI-aims code it was possible to calculate defect concentrations in clathrate networks in agreement with experimental results and to confirm the existence of high temperature phases. As a distinct feature of the calculations, lattice vibrations, determining physical properties towards higher temperatures, were considered thoroughly. The significance of these results goes beyond clathrate research. We expect that the methods may be applied to support complex crystal structure analyses and analysis of structural peculiarities in semiconductor physics.
JG / CPFS

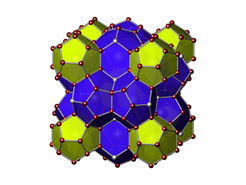
Clathrate-I structure with electropositive metal atoms in the cages and group 14 elements forming the framework. Gray spheres represent possible defect positions.