Zusammensetzung von Clathraten: Strukturanalyse am Computer
Die theoretische Vorhersage und Optimierung von Kristallstrukturen gehört heute zu den Standardwerkzeugen der Materialforschung. Allerdings nimmt man bisher starke Näherungen in Kauf. So sucht man die optimale atomare Anordnung meist bei 0 K unter völliger Vernachlässigung der Entropie, schränkt die Lösungen auf vorgegebene Symmetrien ein, ohne Berücksichtigung von Strukturdefekten. Sind aber zum Beispiel die Halbleitereigenschaften eines Materials von Interesse wie bei thermoelektrischen Materialen, stoßen die etablierten Methoden schnell an ihre Grenzen. In einer Zusammenarbeit des Fritz-Haber-Instituts in Berlin mit dem Forschungsbereich Chemische Metallkunde am MPI CPfS gelang es nun, komplexe strukturelle Details am Beispiel intermetallischer Clathratphasen theoretisch zu berechnen. Die Verwendung des FHI-aims Codes erlaubte es unter anderem, Defektkonzentrationen in Silizium und Germaniumnetzwerken ebenso richtig vorherzusagen, wie die Bildung von Hochtemperaturphasen. Die Besonderheit des Verfahrens liegt darin, dass komplexe Gitterschwingungen einbezogen werden, welche bei höheren Temperaturen die Eigenschaften eines Materials zunehmend beeinflussen. Die Bedeutung der Ergebnisse geht über die Erforschung der Clathrate hinaus, denn es ist zu erwarten, dass das hier verwendete Verfahren komplexe Strukturanalysen in Zukunft erleichtern wird. Dies gilt insbesondere für Halbleiter, deren physikalischen Eigenschaften maßgeblich durch strukturelle Besonderheiten bestimmt werden.
JG / CPFS

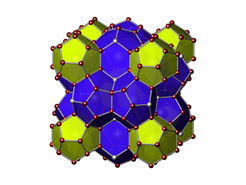
Kristallstruktur einer Clathrat-I Phase mit elektropositiven Metallen in den Käfigen und Gruppe 14 Atomen als Netzwerk. Die grauen Kugeln stellen mögliche Defektpositionen dar.